





Index: Volume 3 Number 1
Next Article: pGATA Positive Selection Vector
 |
Amplification of mRNA Populations Using aRNA Generated from immobilized oligo(dT)-T7 Primed cDNA
Edited from James Eberwine (1996) Biotechniques Vol. 20, No, 4:584-586
Introduction
The amplification of mRNA populations from small amounts of tissue permits the characterization of the expression profile (1) of that tissue. A few years ago we developed the amplified antisense-RNA (aRNA) technology that facilitates the linear amplification of mRNA populations (1). This technique has been successfully used to amplify the mRNA populations of individual cells as well as those present in individual processes of neurons (4,5). A limitation of this methodology is the difficulty in handling small amounts of mRNA and cDNA, where losses can occur in several steps of the procedure and particularly in the phenol/chloroform extraction and the ethanol precipitation steps.
The availability of solid supports for immobilization of nucleic acids has been quite useful in purifying particular RNA populations. Recently oligo(dT) attached to magnetic beads has been produced by several groups (2,3). The main advantage of magnetized beads over cellulose is the easy purification of the oligo(dT)-mRNA complex, which is accomplished by opposing the microcentrifuge tube containing the mixture next to a magnet followed by aspiration of the supernatant.
We have combined the use of magnetic beads with the aRNA amplification procedure to decrease the processing time and to increase the yields in the production of aRNA.
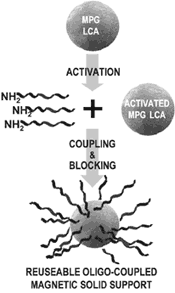 |
Figure 1: Schematic illustration of covalent coupling of 5-NH2 modified oligos to MPG®-LCA beads.The individual steps associated with coupling of the oligo(dT)-T7 primer to the magnetic beads are detailed in the text. |
Results and Discussion
Covalent Coupling of Oligo(dT)-T7 DNA to Magnetic Beads
The solid support used for these studies is the Magnetic Porous Glass (MPG®)-Long Chain Alkylamine (LCA) (Product No. MLCA0502; PureBiotech LLC, Lincoln Park, NJ, USA). This support is magnetized so that the bead and anything attached to it can be manipulated with a magnet. The alkylamine group facilitates the cross-linking of primary amines by activated glutaraldehyde (Figure 1). The amplification oligonucleotide was synthesized with a modified 5' end containing a 6-carbon extension ending with a primary amine 5'NH2-(CH2)6 AAACGACGGCCAGTGAATTGTAATACGACTCACTATAGGCGC- (T)24. The oligonucleotide-MPG® coupling procedure is as follows: Take 1 mL of 10 mg/mL MPG®-LCA and place into a microcentrifuge tube. Use the magnetic separator to aggregate the MPG®-LCA on the side of the tube and remove the aqueous supernatant. Remove the tube from the magnetic separator, add 1 mL coupling buffer and mix. Repeat the magnetic separation procedure. Remove the tube from the separator and add 1 mL 10% glutaraldehyde in coupling buffer (fresh aliquot) and mix well by slowly rotating for 1.5 h at room temperature. Repeat the magnetic separation procedure. Wash the particles with 1 mL of coupling buffer and repeat the magnetic separation procedure. This coupling buffer washing step should be repeated 5 times. One milliliter of coupling buffer containing 1 mg of the oligonucleotide is added to the MPG®-LCA, followed by addition of reducing agent. This is mixed by slow rotation for 5-8 h at room temperature. Repeat the magnetic separation procedure. One milliliter of blocking solution is added to the beads, and this is mixed by slow rotation for 2 h at room temperature. Repeat the magnetic separation procedure. Wash the MPG®-LCA oligonucleotide complex with 1 mL of wash buffer. This washing step should be repeated 5 times. The MPG®-LCA-oligonucleotide is now ready to use.
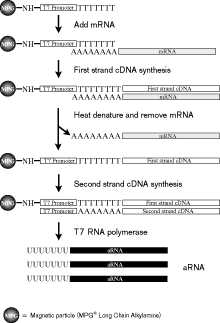 |
Figure 2: Schematic of aRNA synthesis procedure from Immobilized oligo(dT)-T7 primed cDNA.This schematic highlights the different steps associated with synthesis of aRNA from cDNA that has been synthesized directly on the beads. The individual steps associated with each enzymatic reaction are detailed in the text. |
aRNA Synthesis Procedure
The procedure for covalently coupled aRNA synthesis is depicted in Figure 2. This schematic starts at the point of attaching the oligonucleotide covalently to the magnetic beads. Transfer 2-10 µL of the DNA attached beads solution to an RNase-free microcentrifuge tube. Add 10-20 µL of unbound beads (as carrier). Repeat the magnetic procedure to wash the beads twice with 100 µL TE and then equilibrated in cDNA synthesis buffer. The mRNA from a single cell is harvested by using a micropipet to patch directly onto the cell (go into whole cell), followed by aspiration to remove the cellular contents. The micropipet contents are then added directly to the magnetic beads. Mix and incubate at 37°C for 15 min. Add 25 units of reverse transcriptase to the bead solution and incubate at 42°C for 1.5 h. Heat denature the RNA/DNA hybrid by incubating the tube at 90°C for 2-3 min. Cool quickly on ice and magnetically separate and remove the supernatant. The second-strand cDNA synthesis procedure uses random primers to prime the synthesis of second strand cDNA. The conversion of this double-stranded cDNA into aRNA is done using published reaction conditions (4). Figure 3 is a Northern blot showing the size distribution of aRNA synthesized from double-stranded cDNA template that is free in solution compared to that which is covalently coupled to the magnetic beads. In all 5 samples that worked, it can be seen that the size distribution ranges from 500 bases to over 5 kilobases. The size distributions of aRNA are comparable between solution and covalently linked double-stranded cDNA templates.
The beads can be reused at least once to make additional aRNA. It may be possible to reuse them multiple times. If yields decrease with time, probably due to breakdown of the second-strand cDNA, then the double-strand DNA complex should be heat denatured and the second-strand cDNA re-synthesized. This procedure will rejuvenate the cDNA that is attached to the beads. Store the beads at 4°C in storage buffer.
| Figure 3: Comparision of aRNA produt from cDNA in solution and that immpobilized on magnetic beads.This autoradiogram shows the size range of aRNAs made from various cDNA templates electrophoresed on a denaturing 2.5% agarose gel. Lanes 1-3 are the aRNA products of 3 different hippocampal cells from double-stranded cDNA template that was in solution. The sample in Lane 2 did not amplify. Lanes 4-6 are the aRNAs made from cDNAs that were immobilized on the MPG beads. The size distribution shows that the aRNAs range from 600 bases to greater than 5 kilobases for both types of cDNA preparation. | 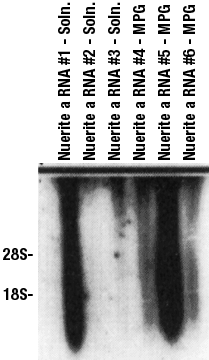 |
The aRNA generated in this procedure can be used for polymerase chain reaction (PCR), expression profiling or as a template for a second round of aRNA amplification using the protocol for aRNA amplification of RNA in solution. It should also be possible to perform the second-round amplification on magnetic beads (since cDNA made from the aRNA will have a poly (A)+ tail), but there is enough RNA at this point that potential losses due to phenol/chloroform extraction and ethanol precipitation of small amounts of nucleic acid should be minimal, although there is still the time consideration.
The conclusions to be drawn from these data are (i) the nucleic acid modification enzymes-reverse transcriptase, E. coli DNA polymerase, T4 DNA polymerase and T7 RNA polymerase-all will function when the substrate is covalently attached to a solid support, and (ii) it is possible to perform aRNA synthesis on an immobilized support. The advantages of coupling the magnetic beads with the oligo(dT)-T7 RNA polymerase primer for the aRNA procedure include speed of performing the procedures, ease of use and an increase of aRNA yield due to the elimination of the phenol/chloroform extraction and ethanol precipitation steps.
1 Eberwine, J., H. Yeh, K. Miyashiro, Y. Cao, S. Nair, R. Finnell, M. Zettel and P. Coleman. (1992). Analysis of gene expression in single live neurons. Proc. Natl. Acad. Sci. USA 89:3010-3014.
2 Hornes, E. and L. Korsnes. (1990). Magnetic DNA hybridization properties of oligonucleotide probes attached to superparamagnetic beads and their use in the isolation of poly(A) mRNA from eucaryotic cells. Genet. Anal. Tech. Appl. 7:145.
3 Jakobsen, K., E. Breivold and E. Hornes. (1990). Purification of mRNA directly from crude plant tissues in 15 minutes using magnetic oligo-dT microspheres. Nucleic Acids Res. 18:3669.
4 Mackler, S., B. Brooks and J. Eberwine. (1992). Stimulus-induced coordinate changes in mRNA abundance in single post-synaptic hippocampal CA1 neurons. Neuron 9:539-548.
5 Miyashiro, K., M. Dichter and J. Eberwine. (1994). On the nature and distribution of mRNAs in hippocampal neurites: implications for neuronal functioning. Proc. Natl. Acad. Sci. USA 91:10800-10804.
James Eberwine
University of Pennsylvania Medical Center
Philadelphia, PA, USA
Next Article: pGATA Positive Selection Vector
© 2004-2023 PureBiotech LLC All rights reserved.