





Index: Volume 2 Number 1
Next Article: Spin Columns
 |
High-Efficiency Full-Length cDNA Cloning by Biotinylated CAP Trapper
Edited from Carninci, P., Kvam, C., Kitamura, A., et al. (1996) Genomics 37: 327- 336.*
Abstract
We have devised a method for efficiently constructing high-content full-length cDNA libraries based on chemical introduction of a biotin group into the diol residue of the cap structure of eukaryotic mRNA, followed by RNase I treatment to select full-length cDNA. The selection occurs by trapping the biotin residue at the cap sites using streptavidin-coated magnetic beads, thus eliminating incompletely synthesized cDNAs. When this method was used to construct a mouse brain full-length cDNA library, our evaluation showed that more than 95% of the total clones were of full length, and recombinant clones could be produced with high efficiency (1.2 x 107/10 µg starting mRNA). The analysis of 120 randomly picked clones indicates an unbiased representation of the starting mRNA population.
Introduction
There are two major technical limitations in full-length cDNA library construction. The first is reduced efficiency of the reverse transcription reaction, which usually cannot efficiently produce full-length first-strand cDNAs, especially when the mRNA presents a stable secondary structure. The second limitation is lack of an efficient technique for selecting only full-length cDNA. Usually, due to reduced representation of full-length clones, several rounds of screening are needed to select the cDNAs carrying the complete sequence. Consequently, conventional methods are time consuming and of low efficiency for selecting the complete recombinant sequence. In this report, we describe a new method for constructing high-content full-length cDNA libraries with efficient production of recombinant full-length cDNA libraries and clones starting from the first transcribed nucleotide. Our method requires only commercially available reagents. For selecting the full-length cDNA, we developed a new technique to label chemically the cap structure with a biotin group, based on the principle that the cap site and 3' end of mRNA are the only sites carrying the diol structure. By using streptavidin-coated magnetic beads, only the full-length first-strand cDNA/mRNA hybrids are selectively recovered after RNase I treatment. The described method also includes a new efficient protocol for synthesizing second-strand cDNA by using only thermostable enzymes. The overall efficiency and yield of the full-length cDNA is thus far superior to other conventional methods. Our method allows the preparation of high-content full-length cDNA libraries, even from relatively small quantities of tissues or early embryos, with no bias in representation since no PCR amplification step has been introduced.
Discussion
The overall methodology is illustrated in Figure 1. In this paper, we describe the development of an efficient method for constructing high-content full-length cDNA libraries that shows four unique features. First, approximately 95% of the clones are of full length, as evaluated by both hybridization and sequencing. The hybridization test of replica filters with probes complementary to the 3' and 5' ends of GAPDH and EF-1-a showed that 98.5 and 95%, respectively, carry both 3' and 5' ends. Additionally, 10 clones of each cDNA were confirmed to be of full length, and random sequencing data showed that about 94% of the clones were of full length. Second, the yield is far higher than those reported with alternative methods. This 10- to 50-fold increase in yield, compared with alternative protocols (1,2), will enable the construction of high-content full-length libraries even from tissues from which only a small amount of mRNA can be prepared, such as early embryos and small organs. Third, the library is free of the bias that can be caused by use of PCR amplification and RNA ligase. Fourth, the protocol can be performed with reagents that are commercially available.
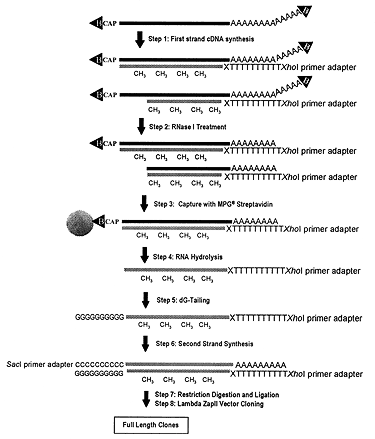
Figure 1.Schematic representation of the general strategy to select and clone full-length cDNA: Step 1, following biotinylation of diol groups, synthesis of hemimethylated first-strand cDNA with MN-degenerate primer adapter; Step 2, protection by full-length cDNA of mRNA from RNase I attack; Step 3, capture of full-length cDNA by streptavidin-coated magnetic beads; Step 4, removal of cDNA from beads by RNase H treatment and mild alkaline RNA hydrolysis; Step 5, addition of oligo(dG) tail to the first-strand cDNA; Step 6, priming of second-strand cDNA synthesis by second-strand primer adapter; Step 7, restriction of the ends of the cDNA with XhoI and SacI; Step 8, cloning of the cDNA in Lambda Zap II.
Since the diol structure is present only on the 5' and 3' ends of mRNAs, we aimed at developing a technique to label the diol group with biotin. Following RNase digestion of the first-strand cDNA reaction, the biotinylated cap can subsequently be used to select only full-length cDNAs. To reach this target, a reaction was developed to label the diol group of the cap structure with biotin by modifying a reaction previously used to biotinylate diol groups of polysaccharides (3,4). The presence of a biotin group on the 3' end of cDNA could lead to an increase of background due to nonspecific capture of the cDNA/mRNA hybrid through the biotin on the 3' end of mRNA. We solved this problem by priming the first-strand cDNA with a short oligo(dT) primer-adapter containing degenerate nucleotides NM at the 3' site, in combination with an optimized annealing program to maximize the priming just at the beginning of the poly(A) (Step 1, Figure 1). Consequently the 3' terminal part of poly(A) was left unprotected from the primer; subsequent addition of RNase I enabled the effective removal of the 3' biotin group (Step 2, Figure 1).
The conditions for the digestion of single-stranded RNA (ssRNA), after first-strand cDNA synthesis, were optimized to remove the cap from the cDNA/mRNA in clones in which first-strand synthesis is incomplete. These conditions allowed protection of the RNA near the cap site from the RNase treatment for only full-length first-strand cDNA. RNase I was selected since it does not show nucleotide specificity (3). Consequently, the cleavage is likely to be effective even for sequences exposing few unprotected nucleotides, as in the case of a complex secondary structure near the 5' site (6), or a very short unprotected segment of the poly(A) end, as for certain mRNAs undergoing degradation (6) (Step 2, Figure 1).
Particular care was taken to minimize the background of the library by selecting optimal reagents. In fact, among other kinds of streptavidin-coated beads, porous glass beads showed the lowest nonspecific adhesion (background) of both unincorporated radioactive deoxynucleotides triphosphate and cDNAs that were not labeled by biotin groups. Additionally, extensive blocking with tRNA was found not to interfere with binding capacity (Step 3, Figure 1).
The synthesis of the second strand was designed to maximize the yield and efficiency of cDNA recombinants. The use of a new temperature gradient was necessary for priming to limit mispairing at the 3' end of the oligo(dG) tail, which is on average a few nucleotides longer than the stretch of 11 C of the primer adapter. Additionally, thermostable enzymes were used to extend effectively even the secondary structure that can be present in the single-stranded cDNA. Ex Taq DNA polymerase was employed because of its proven high performance in long-range PCR (7). Thermostable RNase H and thermostable DNA ligase were added because several stretches of incompletely degraded RNA were likely to remain hybridized to some regions of first-strand cDNA after removal from magnetic beads by partial hydrolysis of the mRNA (Step 4, Figure 1). Those RNA fragments could function as an internal primer for the second-strand synthesis, but must be removed by RNase H treatment to complete the synthesis. Thermostable DNA ligase can join second-strand cDNA fragments that have been internally primed (8). The high yield and efficient production of full-length cDNA can be ascribed to the high efficiency of this step (Steps 5& 6, Figure 1).
We examined whether or not the content of full-length cDNAs would be satisfactory for our applications and superior to those from other methods for full-length cDNA construction. In the oligo-capping method and its modification (1), the contamination rate of truncated clones reaches 30%, which is much higher than that in our library. For Capture method (2), the content of full-length cDNA clones reaches 70%. However, for this method, objective evaluation of data is more difficult, because (a) only 10 clones of one single cDNA encoding for the eIF-4E (2) have been sequenced; (b) eIF-4E shows a stable secondary structure near the 5' end; and (c), the cloning system removed 5' terminal nucleotides, thus ruling out a direct comparison. Additionally, our analysis of two additional libraries produced by omitting the cap trapping confirmed that our protocol was effective in enriching the full-length cDNA content of the library.
More extensive investigations are required to assess if the frequency of clones in the library accurately reflects the original composition of mRNA. However, it is very important that the only bias in our library be due to the different extents to which the reverse transcription can reach the 5' end, since our methodology does not involve other reactions that show sequence specificity like RNA ligation and PCR. By evaluating the average size of the library (1.6 kb) and the high representation of full-length clones, we can roughly estimate that there is a slight preference for cloning short sequences. This is in agreement with the fact that longer cDNAs will be underrepresented due to the limitation of reverse transcriptase reaching the cap site of those mRNAs. Consequently, further improvement in the preparation of the next generation of full-length cDNA libraries may require improvement of the reverse transcription reaction or development of new enzymes. Another possible problem is that some cDNAs whose size exceeds the capacity of the vector may have been lost. There is consequently a need for new vectors with higher cloning capacities for full-length cDNA libraries containing very long clones.
References
-
Details can be obtained by contacting Yoshihide Hayashizaki, Genome Science Laboratory, Tsukuba Life Science Center, The Institute of Physical and Chemical Research (RIKEN), 3-1-1 Koyadai, Tsukuba, Ibaraki 305, Japan Ph: +81 298 36 9145 Fax: +81 298 36 9098.
-
Kato, S., Sekine, S., Oh, S.-W., Kim, N.-S., Umezawa, Y., Abe, N., Yokoama-Kobayashi, M., and Aoki, T. (1994). Construction of a human full-length cDNA bank. Gene 150: 243-250.
Ederly, I., Chu, L.L., Sonenberg, N., and Pelletier, J. (1995). An efficient strategy to isolate full-length cDNA based on an mRNA cap retention procedure (CAPture). Mol. Cell. Biol. 15: 3363-3371.
O'Shannessy, D.J. (1990). Antibodies biotinylated via sugar moieties. Methods Enzymol. 184: 162-165.
Bayer, E.A., and Wilchek, M. (1990). Protein biotinylation. Methods Enzymol. 184: 138-160.
Meador, J., III, Cannon, B., Cannistraro, V.J., and Kennel, D. (1990). Purification and characterization of Escherichia coli RNase I. Comparisons with RNase M. Eur. J. Biochem. 187: 549-553.
Curtis, D., Lehmann, R., and Zamore, P.D. (1995). Translational regulation in development. Cell 81: 171-178.
Cheng, S., Fockler, C., Barnes, W.M., and Higuchi, R. (1994). Effective amplification of long targets from cloned inserts and human genomic DNA. Proc. Natl. Acad. Sci. USA 91: 5695-5699.
Gubler, U., and Hoffman, B.J. (1983). A simple and very efficient method for generating cDNA libraries. Gene 25: 263-269.
Next Article: Spin Columns
© 2004-2023 PureBiotech LLC All rights reserved.